1 产品基本信息
产品名称:One-Step Seamless Cloning Kits
产品编号:MF1808
2 规格或纯度
10 μL×20 T,10 μL×50 T
3 产品介绍
产品简介:
无缝克隆是一种简便、快速且高效的 DNA 定向克隆技术,能将插入片段快速定向克隆到任意载体的任意位点。操作时,先通过任意方法使载体线性化,再在插入片段的正向与反向扩增引物 5' 端引入线性化载体的末端序列,从而让 PCR 产物的 5' 端和 3' 端最末端,分别携带与线性化载体两末端一致的 15-25 bp 序列。随后在重组酶作用下,具有重叠区域的片段与载体可在 50℃条件下最快 5 分钟内完成重组,实现无缝克隆,且该技术的克隆阳性率能达到 95% 以上。具体流程可参见图 1。
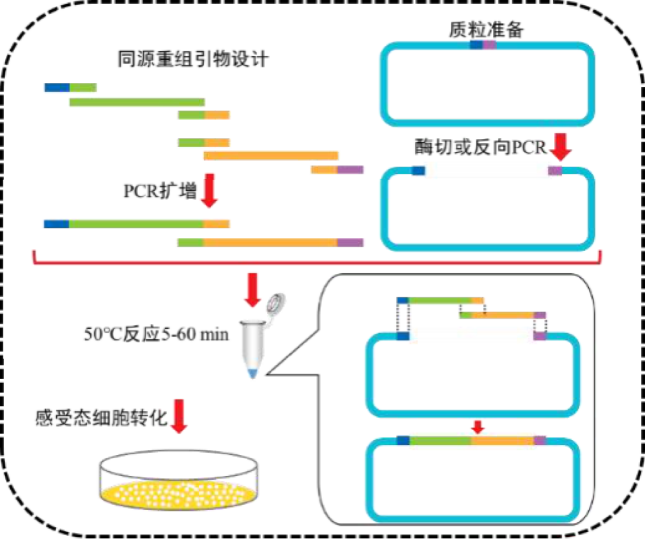
图 1 流程示意图
产品特点:
l操作简便:无需依赖连接酶,不受酶切位点限制,无需对片段进行酶切处理;同时可避免载体自连问题。
l重组高效:单次反应即可完成单个或多个片段的重组,重组过程仅需 5~60 分钟。
l性能优异:支持长片段连接,其效率可与 NEB 无缝克隆试剂盒相媲美。
适用范围:
适用于 1~5 个片段的快速无缝克隆,载体构建、基因合成、定点突变和高通量克隆实验。
产品组分:
组分 | M10701(10 μL × 20 T) | M10701(10 μL × 50 T) |
A. 2× Super Fusion Cloning Mix | 100 μL | 250 μL |
B. Control Plasmid, linearized (Amp+ , 40 ng/μL) | 5 μL | 5 μL |
C. 500 bp Control Fragment (20 ng/μL) | 5 μL | 5 μL |
4 储存与运输
储存条件:-20 ℃ 避光保存;
运输条件:冰袋运输。
5 使用方法(仅供参考)
5.1 制备线性化克隆载体
选择合适的克隆区域对载体进行线性化,此区域的选择应该尽量选择无重复序列且 GC 含量均匀的区域以提高线性化程度,线性化载体可以通过酶切或者反向 PCR 扩增制备。
5.1.1 酶切制备
优先推荐双酶切,此方法会使载体线性化更完全,转化背景(假阳性克隆)低。经双酶切进行线性化的载体无需去磷酸化。
如果使用单酶切,可通过适当延长酶切时间来减少环状质粒的残留。平末端或粘末端均可,但是酶切完全是根本关键。注意,单酶切的线性化载体需要去磷酸化。
注:
(1) 酶切完成后,应将快速内切酶失活或对目的产物纯化后再用于重组反应;
(2) 酶切后需进行胶回收纯化时,可以通过核酸琼脂糖凝胶电泳进行检测产物。
5.1.2 反向 PCR 扩增制备
推荐使用预线性化载体质粒 DNA 为模板,以减少环状质粒模板对克隆阳性率的影响。如果模板为环状质粒时,扩增产物建议使用 Dpn Ⅰ 消化后使用。扩增产物建议使用推荐使用高保真 PCR Mix进行扩增,克隆位点为分界点,设计一对反向引物。扩增产物也可以行胶回收纯化,并通过核酸琼脂糖凝胶电泳进行检测产物。
5.2 设计插入 PCR 引物片段
PCR 引物的 5' 端必须包含与其待重组的相邻片段或载体末端同源的 15~25 nt(推荐 18 nt,不包括酶切位点)序列,使得扩增后的插入片段末端带有和线性化载体末端一致的同源序列。
F 正向引物( 5' -3' ):上游载体重叠区域 + 酶切位点(可选)+ 正向特异性引物扩增序列
F 正向引物( 5' -3' ):下游载体重叠区域 + 酶切位点(可选)+ 反向特异性引物扩增序列
注:
(1) 若载体为粘性末端,且 3' 端突出,则引物设计必须包含部分;若 5' 端突出,则引物设计可以包含突出部分也可以不包含;
(2) 尽量选择无重复序列且 GC 含量均匀的区域进行克隆,当载体克隆位点上下游 25 nt 区域内GC 含量为 40%~60% 时,重组效率最高;
(3) 计算扩增引物 Tm 值时,只需计算特异引物的 Tm 值,引入的额外序列无需计算。
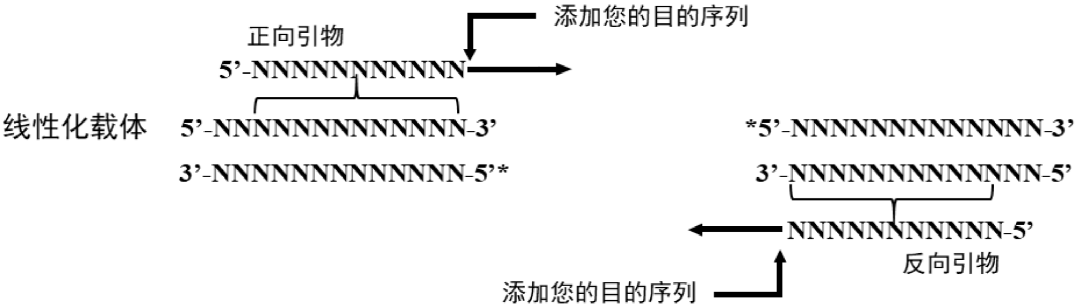
5.2.1 插入片段的 PCR 扩增
插入片段可用任意 PCR 酶(Taq 酶或高保真酶)扩增, 无需考虑产物末端有无 A 尾(重组过程中将被去除,在最终质粒中不会出现)。建议使用高保真聚合酶进行扩增以减少扩增突变的发生。建议使用纯化后的 PCR 产物进行无缝克隆反应,若 PCR 产物经琼脂糖凝胶电泳鉴定为特异性扩增产物,可直接用于无缝克隆反应,但加样的体积不宜超过反应总体积的 20%。
5.2.2 无缝克隆反应
l冰水浴中配制以下反应体系:
组分 | 反应体系 | 阴性对照(1) | 阴性对照(2) | 阳性对照(如有必要) |
2× Super Fusion Cloning Mix | 5 μL | 5 μL | 5 μL | 5 μL |
线性化载体 a | 50~200 ng | 50~100 ng | 1 μL | |
插入片段 b | 10~200 ng | 10~100 ng | 500 bp, 1 μL | |
ddH2O | Up to 10 μL | |||
(a) 最适载体用量(ng)= 0.02 × 载体碱基对数,即 0.03 pmol。
(b) 插入单片段时,最适片段用量(ng)= 0.04 × 片段碱基对数;插入多片段时,每片段最适用量 (ng)= 0.02 × 片段碱基对数。
注:
(1)如果插入的单片段长度大于载体,那么应互换载体与插入片段用量;
(2) 如果插入的片段长度小于 200 bp,那么要使用 5 倍载体的用量;
(3)如果按上述公式计算得到的用量低于最低/高于最高值,那么建议直接按最低/最高用量使用;
(4)载体或插入片段过长,片段数量过多,均会降低阳性率;
(5)阴性对照(1)检验线性化载体中有无背景质粒残留;阴性对照(2)当插入片段扩增模板是与克隆载体抗性相同的环状质粒时, 推荐进行;
(6)若某一组分浓度过高可适当稀释后使用,每一个组分的用量最好不低于 1 μL;
(7)体系配制完成后,轻轻吹吸数次混匀各组分,避免产生气泡即可,切勿涡旋。
l将反应体系置于 50 ℃,反应 5~60 min。
注:
(1) 推荐使用温控比较精准的仪器进行反应,如 PCR 仪,反应时间不足或过长都会降低克隆效率;
(2) 插入 1~2 个片段时,推荐反应时间为 5~15 min;插入 3~5 个片段时,推荐反应时间为 15~30
min;
(3) 当载体骨架在 10 kb 以上或插入片段在 4 kb 以上时,建议延长反应时间到 30~60 min;
(4) 50 ℃ 反应完成后,建议进行瞬时离心,将反应液收集至管底。
l 将反应液离心管置于冰水浴中冷却,建议即时转化或储存于 -20 ℃。
注:-20 ℃ 储存的重组产物,建议在 1 周内使用。
5.3 克隆产物转化
在 100 μL 感受态细胞中加入 5~10 μL 反应液(体积不超过所用感受态细胞体积的 1/10),轻轻混匀,置于冰上 25 min。然后 42 ℃ 热激 45~60 s,迅速转入冰浴中静置 2- 5 min。加入 500~700 μL SOC 或LB 培养基(不含抗生素的无菌培养基),混匀后 37 ℃ 振荡培养 40~60 min(200 rpm)。根据实验需要,将菌液均匀涂布在含相应抗生素的平板上,将平板倒置于 37 ℃ 培养箱培养过夜。
注:
(1) 不同感受态细胞最后的克隆阳性率会有所差别,推荐使用转化效率 > 108 cfu/μg 的感受态细胞;
(2) PCR 产物与线性化载体的数量和纯度决定了菌落数;
(3) 阳性对照平板通常生长大量白色单菌落,阴性对照平板只生长很少的菌落。
5.4 阳性克隆检测
5.4.1 菌落/菌液 PCR 鉴定:
挑取单菌落至 10 μL ddH2O 中混匀,取 1 μL 作模板,或直接蘸取菌落进行菌落 PCR 鉴定(建议菌落 PCR 时,至少使用一条通用引物,可有效避免假阳性结果);
5.4.2 以质粒为模板 PCR 鉴定:
挑取单克隆至含相应抗生素的 LB 培养基中,37 ℃, 200 rpm 过夜摇菌后抽提质粒作为模板,可使用载体通用引物或特异性引物扩增;
5.4.3 酶切鉴定(若有需要):
挑取单克隆至含相应抗生素的 LB 培养基中,37℃, 200 rpm 过夜摇菌后抽取质粒,使用相应内切酶切质粒后电泳检测片段大小。
注:
(1) 必要时可进一步对阳性结果进行测序鉴定;
(2) 测序引物序列:M13F:TGTAAAACGACGGCCAGT;M13R:CAGGAAACAGCTATGAC;
6 注意事项
l本产品仅限于科研用途并且不得存放于普通住宅内。
l为了您的安全和健康,请遵循您所在常规实验室安全规定。
(1) 测序的样本数:如果只测了一个样本,不一定可信,需要多测几个样本;
(2) 分析测序结果:检查突变处的测序信号是否有问题,若为双峰或者杂峰则不一定可信;
(3) 检查突变位点:如果多个结果突变位点一致,可能是从模板上引入的或者模板序列与 NCBI上不一致;
(4) 插入片段在 PCR 获得时建议使用高保真聚合酶;
(5) 若引物处发生碱基突变,考虑是引物合成问题,建议重新合成引物进行实验。
(1) 质粒载体残留导致的假阳性:设置阴性对照来排查,如果阴性对照长了很多,可能是模板投入量过多或者未进行 Dpn Ⅰ 消化。若以质粒为模板 PCR 获得片段/载体:质粒投入量不宜过多(<1 ng),否则可能导致 Dpn Ⅰ 消化不完全从而造成假阳性。建议 PCR 产物进行凝胶回收纯化,纯化产物溶解在 ddH2O 中;
(2) 挑斑方法不合适:重组产物 3' 端与 5' 端未连接,需要到感受态细胞中进行修复,斑点相对来说会小点;平板营养不均匀会导致斑点大小有差异;重组质粒相对原始质粒来说,有可能会影响细胞的生长。建议选取平板上某一块区域,同时挑选大斑和小斑,以增加挑到重组产物的概率;
(3) 菌检引物不合适:建议使用载体通用引物或者引物一端在载体上,一端在目的片段上。如果用目的片段引物进行菌液 PCR,可能会出现菌检有结果而测序无结果。
(1) 相同抗性质粒污染。PCR 扩增模板为环状质粒时,如扩增产物未纯化直接用于重组反应时推荐 Dpn Ⅰ 消化,或者对扩增产物进行胶回收纯化;
(2) 克隆载体及片段中含有质粒背景。克隆载体线性化不完全,酶切制备线性化载体时,要提高快速内切酶的使用量并延长反应时间,使用胶回收纯化酶切产物;制备插入片段时尽量⽤预线化质粒作为扩增模板,扩增产物进行 Dpn Ⅰ 消化以及进行凝胶回收纯化;
(3) PCR 产物混有非特异扩增产物:优化 PCR 体系,提高特异性;胶回收 PCR 产物;鉴定更多的克隆。
(1) 载体线性化不完全,没有完全切开;
(2) 目的片段投入过低,导致连接失败。
(1) 感受态细胞因为保存时间或者质量不佳会引起转化效率低下, 建议使用新制备的感受态细胞或者质量上佳的感受态细胞;
(2) 线性化载体或插入片段比例不佳。请严格按照说明书推荐的方法计算各组分用量,用琼脂糖凝胶电泳检测样品质量以及浓度;
(3) EDTA 等金属离子螯合剂会抑制无缝克隆实验;
(4) 胶回收制备,最后洗脱时需要用水洗脱,不能用 TE Buffer 进行洗脱,用于重组反应的胶回收产物要溶解于 ddH2O;
(5) 插入片段纯度差;
(6) 引物设计有误,设计时特别注意反向引物的序列顺序。


关注公众号

关注视频号
合作咨询
扫描二维码
咨询合作
联系销售
7X24小时客服
0512-66523352
公众号
顶 部